CANALOPATIAS.
Son trastornos eléctricos primarios (eléctricos puros), sin que exista cardiopatía estructural, causados por mutaciones congénitas de proteínas que actúan como canales iónicos trasmembrana.
1 SINDROME DE BRUGADA (SBr).
.- Prevalencia sub-registrada de 1/2.000 habitantes.
.- En Asia es llamado Sd de muerte súbita inesperada nocturna y se relaciona con el Sd de muerte súbita del lactante.
.- Mutación autosómica dominante y de novo de:
- Subunidad alfa del canal de Na+ dependiente de voltaje: que causa perdida de su función llevando a una disminución en la entrada de Na+ a la célula al final de la fase 0 del potencial de acción.
- Canales de Ca++ tipo L: que causa perdida de su función llevando a una disminución en la entrada de Ca++ a la célula en la fase 1 y 2 del potencial de acción.
- Canales de K+ dependientes de voltaje: Aumentando la corriente de salida de K+ de la célula en la fase 1 del potencial de acción.
.- Fisiopatología del SBr.
- La disminución de las corrientes positivas de entrada y/o el incremento de las corrientes positivas de salida resultan en una acentuación de la muesca al final de la fase 1 del potencial de acción, lo cual causa una repolarización muy rápida, lo cual se manifiesta en el EKG con una supradesnivelación del ST (porque la repolarización es muy rápida y no alcanza a llegar a la línea isoeléctrica).
- La exageración de los cambios iónicos puede provocar un gran acortamiento o incluso pérdida de la meseta de la fase 2 del potencial de acción, aumentándose aún más la velocidad de repolarización llevando a un periodo supernormal de la fase 4 del potencial de acción muy rápidamente.
- En ocasiones se presenta dispersión de la repolarización en diferentes zonas miocárdicas, o sea que se presenten zonas donde se encuentre en periodo supernormal excitable de la fase 4 y otras donde aún esté en proceso de despolarización (donde la repolarización no es tan acelerada). La propagación del estímulo eléctrico desde zonas donde la meseta de la fase 2 está mantenida hacia zonas del miocardio donde ha desaparecido o está muy disminuida, causa reexcitación mediante fenómeno de reentrada, que causa la aparición de extrasístoles ventriculares que van a desencadenar la TVP o la FV y esta lleva a la MS.
.- Las arritmias y la MS se presenta más frecuentemente a los 40 años durante el reposo, sueño o luego de una comida copiosa.
.- Sd que se presenta con:
- Patrón de BRDHH,
- Elevación persistente del segmento ST.
- Ausencia de cardiopatía estructural.
- Predisposición a presentar taquicardia ventricular polimorfa y fibrilación ventricular que lleva a muerte súbita –MS- (causa hasta el 20% de las muertes súbitas).
.- Se han descrito 3 tipos con trazados EKG característicos:
- Tipo 1 de SBr: Elevación de ST (>2mm) + onda T negativa en precordiales derechas (V1-V2 – V3) y en ocasiones en inferiores (DII, aVF, DIII)
- Tipo 2 de SBr: Elevación de ST + onda T positiva o en “silla de montar” en precordiales derechas (V1-V2 – V3) y en ocasiones en inferiores (DII, aVF, DIII)
- Tipo 3 de SBr: morfología cóncava o convexa, con elevación del segmento ST (<1 mm) se consideran sugestivos, aunque no diagnósticos de la patología.
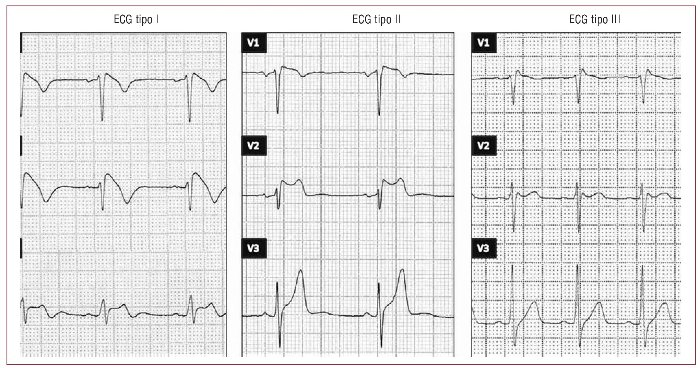
El diagnóstico sólo debe establecerse cuando el patrón ECG tipo I se asocia al menos a una de los siguientes criterios clínicos:
- Fibrilación ventricular (FV),
- Taquicardia ventricular polimórfica (TVP),
- Inducibilidad de arritmias ventriculares durante el estudio electrofisiológico (EEF),
- Síncope o respiración agónica nocturna,
- Historia familiar de MS en edad previa a los 45 años o
- Patrón ECG tipo I en otros miembros de la familia
2 SINDROME DE QT LARGO (SQTL).
Son un tipo de canalopatía congénitas, que se caracteriza por trastornos de la repolarización, con dispersión de la refractariedad y con posdespolarizaciones que producen taquiarritmias, que se manifiestan por sincope y muerte súbita, y se clasifican desde el 1 hasta el 13.
2.1 SQTL tipo 1:
.- Mutación de la subunidad alfa de la proteína del canal de la corriente lenta rectificadora de K de la fase 3 del potencial de acción, permaneciendo cerrados, acumulando K intracelularmente lo cual prolonga la repolarización y por tanto el QT.
.- Es la más frecuente (50% de los casos).
.- Se desencadena la arritmia con el ejercicio o estimulación adrenérgica.
.- EKG: onda T muy ancha y alta (duración y amplitud).
.- El potencial de acción se prolonga por una disminución de la corriente saliente de K+ durante fase 3 del potencial de acción.
.- Este canal de IKs también se encuentra en la estría vascularis del oído interno controlando la producción de linfa, por esta razón los paciente con este síndrome tipo 1 se acompañan de sordera.
2.2 SQTL tipo 2:
.- Mutación de la subunidad alfa de la proteína del canal de la corriente rápida rectificadora de K de la fase 3 del potencial de acción, permaneciendo cerrados, acumulando K intracelularmente lo cual prolonga la repolarización y por tanto el QT.
.- Es la segunda más frecuente (30% de los casos).
.- Se desencadena la arritmia con la estimulación adrenérgica, estímulos sonoros súbitos (despertador, alarmas) y menos frecuente con el sueño y el ejercicio.
.- EKG: onda T muy ancha bífida pero baja (larga duración y poca amplitud).
.- El potencial de acción se prolonga por una disminución de la corriente saliente de K+ durante fase 3 del potencial de acción.
2.3 SQTL tipo 3:
.- Mutación de la subunidad alfa del canal de Na+ dependiente de voltaje, permaneciendo abierto y por tanto esta inactivación defectuosa en la fase 1 del potencial de acción permite una entrada sostenida de Na+ prolongando su duración, alargando el segmento ST de la repolarización.
.- Causa del 5 al 10% de los casos.
.- Se desencadena arritmias malignas durante el reposo (sueño), siendo menos sintomáticos pero más letales.
.- EKG: Segmento ST largo con onda T pequeña, picuda y tardía (acuminada).
.- El potencial de acción se prolonga por entrada sostenida de Na+ en la fase 2 del potencial de acción.
2.4 SQTL tipo 4:
.- Raro (1% de los casos).
.- Mutación de la proteína anquirina B (inactivándola), proteína que tiene por función vincular proteínas esenciales de la membrana al citoesqueleto del miocardiocito como son la bomba Na+/ K+ ATPasa, proteína intercambiadora de Na+/Ca++ y el receptor del IP3, mutación que causa la perdida de la funcionalidad de estas proteínas, llevando a la acumulación de Na+ y Ca++ intracelularmente llevando a posdepolarizaciones tempranas y tardías, por aumento de la positividad eléctrica intracelular.
.- Desencadena un gran espectro de arritmias tales como disfunción del nodo sinusal, fibrilaciones auriculares, trastornos de la conducción AV y la taquicardia ventricular polimorfa catecolaminérgica.
.- El potencial de acción se prolonga por altas concentraciones de Na+/Ca++ intracelular.
2.5 SQTL tipo 5:
.- Muy raro (menos 1% casos).
.- Todo es igual al tipo 1 pero la subunidad mutada es la beta del canal lento de K.
2.6 SQTL tipo 6:
.- Muy raro (menos 1% casos).
.- Todo es igual al tipo 2 pero la subunidad mutada es la beta del canal rápido de K.
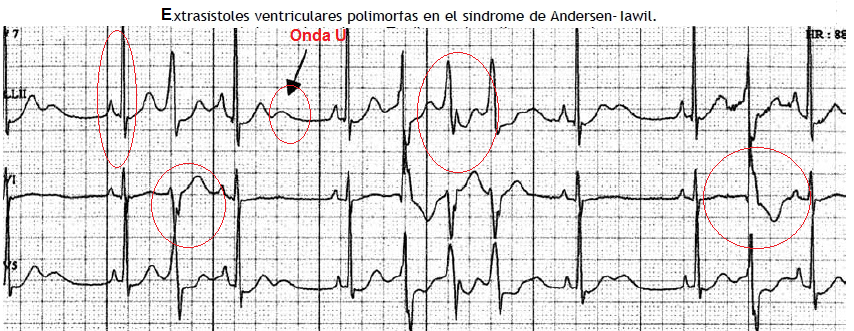
2.7 SQTL tipo 7 o Andersen-Tawil.
.- Mutación del canal rectificador de K de la fase 4 del potencial de acción, que impide la repolarización normal por aumento del K intracitoplasmático.
.- Es un Sd con alteraciones:
- Arritmias ventriculares: extrasístoles ventriculares, taquicardia ventricular bidireccional o dismórfica, taquicardia ventricular por torsión de puntas hasta fibrilación ventricular.
- Alteraciones musculoesqueléticas y dismorfismo: hipertelorismo, micrognatia, implantación baja de pabellón auricular, talla baja, escoliosis, clinodactilia.
.- EKG: El QTc puede estar normal o ligeramente aumentado, pero hay onda u prominente.
.- Es un diagnóstico diferencial de la taquicardia ventricular polimorfa catecolaminérgica, de la cual se diferencia en que los complejos dismórficos se presentan en reposo.
2.8 SQTL tipo 8 o Sd de Timothy:
.- Muy raro (0,5% de los casos).
.- Mutación de los canales de calcio tipo L de la fase 2 del potencial de acción.
.- Es un Sd con alteraciones:
- Malformaciones cardiacas-
- Arritmias cardiacas con QT largo y muerte súbita.
- Deficiencias inmunológicas.
- Hipoglicemia.
- Trastornos cognitivos incluyendo autismo.
2.9 SQTL tipo 9
.- Rara (1% de los casos).
.- Mutación de la proteína caveolina 3, la cual es una proteína necesaria para la formación de invaginaciones en la membrana celular que participan en la endocitosis de lípidos y neurotransmisores. Existen 3 isoformas de la caveolina, siendo la isoforma 3 específica del músculo estriado voluntario e involuntario (cardiaco) y en las invaginaciones qué forma se localiza los canales de Na+ dependientes de voltaje; por tanto al estar mutada y no formarse adecuadamente las invaginaciones se alteran los canales de Na+ dependientes de voltaje de la fase 1 del potencial de acción y por tanto se manifiesta este SQTL 9 igual que el SQTL3.
2.10 SQTL tipo 10
.- Rara (<1% de los casos).
.- Mutaciones de la subunidad β 4 de canal de sodio dependiente de voltaje.
3 SINDROME DE QT CORTO (SQTC).
Esta es otra canalopatía también congénita, que se presenta por mutación en los canales rápidos de Potasio, los cuales son los que llevan a cabo la corriente rápida de recuperación de K en la fase 3 del potencial de acción, denominados canales IKf (la f es de fast en inglés), estos canales mutados permanecen abiertos , permiten la salida más rápida del K intracelular, lo cual trae como consecuencia una drástica disminución del tiempo de repolarización o incluso acelera este tiempo, lo cual hace que sea más propenso a arritmias incluyendo la fibrilación auricular; o sea este síndrome de QTC es lo contrario a los SQTL tipo 2.
En el SQTC el QT corregido tiene menos de 360 ms (sin embargo es muy sintomático cuando es menor de 320 ms), que se acompaña de arritmias ventriculares malignas y llevan a muerte súbita, de los cuales hasta la fecha se han descrito 5 tipos.
El QT sea acorta además en los siguientes casos:
- Fisiológicamente en el aumento de la frecuencia cardiaca.
- Hipercalcemia.
- Hipercalemia (K en el espacio intersticial).
- Hipertermia.
- Acidosis.
- Administración de catecolaminas y acetilcolina.
Los pacientes con QT corto de tipo congénito presentan el QT corto incluso en bradicardias.
Criterios electrocardiográficos:
a) Obviamente QT corto, menor de 360 ms.
b) Ondas T altas, picudas, de base estrecha (porque el QT está corto) y simétrica, las cuales incluso pueden alcanzar la misma altura que los QRS. Recordemos que unas ondas T de similares características se presentan en la isquemia aguda subepicárdica, hiperkalemia, S. de repolarización precoz, pericarditis, alcoholismo, bloqueo de rama izquierda, en las sobrecarga diastólica de ventrículo izquierdo y en la vagotomía.